Abstrak
Kemajuan terkini dalam perangkat keras dan perangkat lunak telah memungkinkan simulasi dinamika molekuler (MD) biomolekul yang semakin lama, mengungkap keterbatasan tertentu dalam akurasi medan gaya yang digunakan untuk simulasi tersebut dan memacu upaya untuk menyempurnakan medan gaya ini. Modifikasi terkini pada medan gaya protein Amber dan CHARMM, misalnya, telah meningkatkan potensi torsi tulang punggung, memperbaiki kekurangan pada versi sebelumnya. Di sini, kami lebih jauh memajukan akurasi simulasi dengan meningkatkan potensi torsi rantai samping asam amino dari medan gaya Amber ff99SB. Pertama, kami menggunakan simulasi sistem alfa-heliks model untuk mengidentifikasi empat jenis residu yang distribusi rotamernya paling berbeda dari ekspektasi berdasarkan statistik Protein Data Bank. Kedua, kami mengoptimalkan potensi torsi rantai samping residu ini agar sesuai dengan kalkulasi mekanika kuantum tingkat tinggi yang baru. Akhirnya, kami menggunakan simulasi MD skala waktu mikrodetik dalam pelarut eksplisit untuk memvalidasi medan gaya yang dihasilkan terhadap serangkaian besar pengukuran NMR eksperimental yang secara langsung menyelidiki konformasi rantai samping. Medan gaya baru, yang kami sebut Amber ff99SB-ILDN, menunjukkan kesesuaian yang jauh lebih baik dengan data NMR. Proteins 2010. © 2010 Wiley-Liss, Inc.
PERKENALAN
Simulasi dinamika molekul (MD) dengan medan gaya atomistik berbasis fisika menawarkan potensi untuk mendapatkan wawasan baru ke dalam mekanisme fungsional biomolekul. Namun, keberhasilan penerapan simulasi tersebut mungkin dibatasi oleh dua kekurangan utama. Pertama, persyaratan komputasi membatasi jumlah waktu yang dapat disimulasikan dan dengan demikian dapat mencegah simulasi dari mengeksplorasi semua konformasi molekuler yang relevan ( masalah pengambilan sampel ). Kedua, ketidakakuratan dalam fungsi energi potensial dapat membiaskan simulasi ke arah konformasi yang salah ( masalah medan gaya ). 1 Meskipun kemajuan dalam mengatasi masalah pengambilan sampel dapat membuat fenomena biologis baru dan penting dapat diakses untuk studi komputasi untuk pertama kalinya, keberhasilan upaya tersebut sangat bergantung pada kualitas medan gaya, karena ketidakakuratan dalam model fisik yang digunakan untuk simulasi molekuler dapat menghasilkan hasil yang menyesatkan bahkan tanpa adanya batasan komputasi. Kemajuan terbaru dalam perangkat lunak dan perangkat keras telah memungkinkan simulasi proses yang relevan secara biologis dengan akurasi atomistik pada skala waktu yang jauh melampaui mikrodetik. 1 – 3 Perkembangan ini, dipadukan dengan perbaikan berkelanjutan pada teknik pengambilan sampel yang lebih baik, 4 telah menimbulkan tuntutan yang semakin besar pada akurasi medan gaya.
Seperti yang dicontohkan oleh penelitian terbaru tentang asam nukleat 5 dan protein, 2 simulasi MD panjang telah digunakan untuk menyoroti kekurangan dalam medan gaya yang ada, yang pada gilirannya mengarah pada pengembangan versi yang baru dan lebih baik. 5 Meskipun banyak pengembangan saat ini di area ini difokuskan pada penyertaan efek polarisasi, 6 medan gaya yang dapat dipolarisasi secara komputasi lebih mahal daripada medan gaya bermuatan tetap, dan penelitian terbaru menunjukkan bahwa masih ada ruang untuk perbaikan medan gaya yang tidak dapat dipolarisasi. Modifikasi kecil namun penting pada potensi torsi tulang punggung yang dimasukkan dalam versi terbaru medan gaya Amber dan CHARMM (Amber ff99SB 7 dan koreksi energi tulang punggung CMAP ke CHARMM22 8 ) telah menghasilkan peningkatan akurasi dibandingkan dengan rilis sebelumnya, seperti yang ditunjukkan, misalnya, melalui perbandingan hasil simulasi dengan data eksperimen. 9
Di sini, kami lebih menyempurnakan medan gaya protein Amber ff99SB dengan mengoptimalkan potensi torsi χ 1 untuk rantai samping asam amino. Di antara derajat kebebasan torsi dalam protein, torsi χ 1 diperkirakan menjadi yang kedua setelah torsi tulang punggung dalam hal pentingnya untuk menggambarkan energi protein, namun istilah yang relevan dalam medan gaya Amber tetap hampir identik selama 25 tahun. 10 – 12 Karena potensi torsi χ 1 ini , menurut pengetahuan kami, belum direvisi secara sistematis sejak diperkenalkan pertama kali, hal itu tampak sebagai target alami untuk perbaikan.
Di sini kami memfokuskan upaya kami pada rantai samping yang menampilkan penyimpangan terbesar dari perilaku yang diharapkan dan menggunakan prosedur tiga langkah untuk menyempurnakan medan gaya. Pertama, kami mengidentifikasi jenis residu yang dianggap bermasalah dengan membandingkan distribusi dihedral χ 1 dalam simulasi peptida heliks pendek dengan statistik yang sesuai untuk residu dalam heliks di Protein Data Bank (PDB). 13 Kami menemukan bahwa empat jenis residu (isoleusin, leusin, aspartat, dan asparagin) menunjukkan penyimpangan yang sangat besar dari distribusi PDB, yang menunjukkan bahwa medan gaya ff99SB tidak memodelkan rantai samping ini dengan baik. Kedua, kami memperoleh potensi torsi χ 1 baru untuk keempat residu ini dengan menyesuaikan parameter medan gaya ke pemindaian dihedral DF-LMP2 14 , 15 tingkat kuantum ab initio . Akhirnya, kami memvalidasi medan gaya yang disempurnakan menggunakan sekumpulan besar data NMR yang berisi ratusan pengukuran yang secara langsung menyelidiki konformasi rantai samping yang relevan. Kami menemukan peningkatan substansial untuk keempat residu yang dimodifikasi, seperti dibuktikan oleh kesesuaian yang jauh lebih dekat antara keadaan rotamerik yang diamati dalam simulasi dan yang diuji oleh eksperimen NMR.
BAHAN DAN METODE
Simulasi MD heliks polialanin pendek
Kami melarutkan heliks berbasis alanin yang ditutup secara terminal dengan urutan Ace-(Ala) 4 Xaa(Ala) 4 -NMA, di mana Xaa adalah salah satu dari 20 asam amino yang terjadi secara alami selain Gly, Ala, dan Pro, dalam kotak kubik dengan sisi ∼27Å yang berisi ∼600 molekul air. Keadaan protonasi dipilih agar sesuai dengan pH netral. Karena tujuannya adalah untuk membandingkan distribusi rotamer yang diamati dalam simulasi MD peptida ini dengan distribusi yang diamati dalam heliks, kami menerapkan pengekangan lemah pada sudut torsi ϕ dan φ untuk memastikan bahwa peptida tetap berbentuk heliks. Pengekangan ini berbentuk:
dengan nilai referensi (θ 0 ) sebesar 122° dan 133° untuk ϕ dan φ, masing-masing, dan konstanta gaya ( k θ ) sebesar 1 kcal mol −1 . Kami mencatat bahwa meskipun nilai referensi tidak sesuai dengan daerah heliks dari peta Ramachandran, deret kosinus ini bertindak sebagai pengekangan yang memastikan bahwa peptida tetap dalam konformasi heliks di seluruh simulasi tanpa secara nyata mempengaruhi gerakan rantai samping.
Setiap sistem diseimbangkan pada 300 K dan 1 atm dengan simulasi MD 2,4 ns dalam ensembel NPT. Kemudian, simulasi MD dilakukan dalam ensembel NVT selama 720 ns menggunakan termostat Nosé-Hoover dengan waktu relaksasi 1 ps. Semua simulasi dilakukan menggunakan program Desmond MD versi 16 2.1.1.0 dan Amber ff99SB 7 atau medan gaya Amber ff99SB yang dimodifikasi yang dijelaskan di sini, yang kami sebut ff99SB-ILDN. Semua ikatan yang melibatkan atom hidrogen dibatasi dengan algoritma SHAKE. 17 Batas 10 Å digunakan untuk interaksi Lennard-Jones dan interaksi elektrostatik jarak pendek. Metode jaring partikel halus Ewald 18 dengan kisi 32 × 32 × 32 dan skema interpolasi orde keempat digunakan untuk menghitung interaksi elektrostatik jarak jauh. Daftar pasangan diperbarui setiap 10 fs dengan batas 10,5 Å. Kami menggunakan skema RESPA multilangkah 19 untuk integrasi persamaan gerak dengan langkah waktu 2,0, 2,0, dan 6,0 fs untuk interaksi terikat, tidak terikat jarak pendek, dan tidak terikat jarak jauh, masing-masing. Untuk memeriksa potensi bias yang disebabkan oleh interaksi jarak jauh antara peptida dalam gambar periodik, kami mengulangi simulasi ini untuk empat asam amino (Xaa: Ile, Leu, Asp, dan Asn) menggunakan kotak yang lebih besar dengan panjang sisi 37 Å. Kami menemukan bahwa hasil simulasi kontrol ini berada dalam kesalahan yang sama dengan hasil simulasi yang menggunakan ukuran kotak yang lebih kecil.
Simulasi MD protein globular kecil
Simulasi MD lisozim putih telur ayam (HEWL), penghambat tripsin pankreas sapi (BPTI), ubikuitin (Ubq), dan domain B3 Protein G (GB3) dilakukan menggunakan Desmond versi 2.1.0.1 dan medan gaya Amber ff99SB atau Amber ff99SB-ILDN yang dimodifikasi. Model air TIP3P 20 digunakan untuk simulasi HEWL, Ubq, dan GB3, dan model air TIP4P-Ew 21 digunakan untuk simulasi BPTI. Parameter simulasi sama seperti dalam simulasi peptida heliks kecil, selain fakta bahwa kisi 64 3 PME digunakan untuk HEWL dan kisi 48 3 digunakan untuk BPTI, Ubq, dan GB3. Simulasi HEWL, BPTI, Ubq, dan GB3 dimulai dari PDB 22 entri 6LYT, 5PTI, 1UBQ, dan 1P7E yang dilarutkan dalam kotak air kubik yang masing-masing berisi 10.594, 4215, 6080, dan 5156 molekul air. Muatan bersih protein dinetralkan dengan ion natrium atau klorida. Setiap sistem awalnya mengalami minimisasi energi, diikuti oleh 1,2 ns simulasi MD dalam ansambel NPT yang selama itu suhu dinaikkan secara linear dari 10 hingga 300 K, dan pengekangan posisi pada atom-atom tulang punggung dianil dari 1,0 hingga 0,0 kkal mol −1 Å −1 . Setelah relaksasi awal ini, setiap sistem disimulasikan selama 6 ns dalam ansambel NPT. Kerangka simulasi ini dengan volume yang paling mendekati volume rata-rata dipilih sebagai konformasi awal untuk uji coba produksi 1,2 μs dalam ensembel NVT. Lintasan yang diperoleh dari uji coba NVT digunakan untuk analisis data selanjutnya.
Perhitungan sifat NMR
Untuk keempat sistem protein, konstanta kopling 3 J yang diukur secara eksperimental untuk dihedral H α –C α –C β –H β1,2 tersedia 23 – 27 (dan Bax, komunikasi personal). Nilai eksperimental dibandingkan dengan yang dihitung menggunakan hubungan Karplus 28 dari sudut torsi yang diamati dalam simulasi MD. Untuk BPTI, HEWL, dan Ubq, penugasan stereo-spesifik memungkinkan kita untuk membedakan antara kopling untuk H β1 dan H β2 . Dalam GB3, di mana penugasan stereospesifik tidak tersedia, kami menggunakan kopling dipolar residual (RDC) C β –H β1,2 yang diukur secara independen untuk menentukan penugasan yang paling mungkin, seperti yang telah disarankan sebelumnya. 26 Selain menghitung kopling H α –C α –C β –H β1,2 untuk keempat protein, kami juga menghitung kopling N–C α –C β –C γ dan C′–C α –C β –C γ dalam GB3 dan Ubq 29 , 30 dan kopling C′–C α –C β –H β1,2 dalam HEWL. 24 Untuk kopling N–C α –C β –C γ dan C′–C α –C β –C γ dalam Ile, Val, dan Thr, kami menggunakan parameter Karplus dari Chou et al . 30 ; untuk semua kopling lainnya, kami menggunakan parameter spesifik asam amino dari Pérez et al . 31
RDC rantai samping untuk GB3 dan HEWL dihitung sebagai rata-rata ensemble, seperti yang dijelaskan sebelumnya. 32 Untuk HEWL, tensor penyelarasan pertama-tama ditentukan menggunakan satu set RDC HN backbone, dan tensor penyelarasan yang dihasilkan kemudian digunakan untuk menghitung RDC untuk amida rantai samping Asn. 33 Karena percobaan hanya melaporkan jumlah dari dua RDC untuk ikatan N δ –H δ1,2 , kami menghitung jumlah yang sama dari simulasi. Dalam GB3, prosedur yang sama digunakan untuk menentukan tensor penyelarasan dari satu set kopling backbone, yang menghasilkan perhitungan kopling C β –H β1,2 . 26 Secara total, 390 kopling skalar dan 50 RDC dihitung dari simulasi MD dan dibandingkan dengan nilai eksperimen. Set data lengkap, bersama dengan nilai yang dihitung menggunakan ff99SB dan ff99SB-ILDN, tersedia dalam Informasi Pendukung untuk artikel ini.
Perhitungan ab initio
Perhitungan mekanika kuantum (QM) dilakukan pada level teori MP2, menggunakan perkiraan lokal dan perkiraan kepadatan, 14 dengan set basis triple-zeta yang diperbesar (aug-cc-pVTZ) melalui program MOLPRO. 15 Pemindaian penuh permukaan energi potensial (PES) di sekitar ikatan χ 1 dilakukan untuk peptida Ace-Xaa-NMA, di mana Xaa adalah Ile, Leu, Asp, atau Asn. Untuk setiap titik pada PES, geometri sistem sepenuhnya direlaksasikan dengan sudut χ 1 , χ 2 , ϕ, dan φ dibatasi. Dalam perhitungan Ile dan Leu, χ 1 divariasikan antara −180° dan 180° dalam kenaikan 15°, dan untuk setiap nilai χ 1 , χ 2 nilai −60°, 60°, dan 180° dipertimbangkan. Sebanyak 72 titik dioptimalkan untuk masing-masing dari dua residu ini. Untuk Asp dan Asn, baik χ 1 maupun χ 2 divariasikan antara −180° dan 180° dalam kenaikan 30°. Sebanyak 72 dan 144 titik dioptimalkan untuk Asp dan Asn, masing-masing (perhatikan bahwa perhitungan peta torsi Asp χ 1 /χ 2 memerlukan setengah jumlah titik karena simetri torsi χ 2 di Asp). Dalam semua perhitungan, ϕ dan φ dijaga tetap pada nilai masing-masing -135° dan 135°, yang sesuai dengan konformasi yang diperluas.
Penyesuaian parameter
Kesesuaian dengan pemindaian energi potensial dilakukan dengan menghitung perbedaan antara energi mekanika molekuler dan energi ab initio untuk setiap titik pada PES. Istilah energi torsi χ 1 di Ile dan Leu serta torsi χ 1 dan χ 2 di Asp dan Asn kemudian dioptimalkan untuk meminimalkan fungsi berikut:
di mana E MM dan E QM masing-masing adalah energi medan gaya dan QM, dan N adalah jumlah konformasi yang dioptimalkan pada tingkat QM. Perbedaan antara E MM dan E QM dibobot dengan faktor Boltzmann gambar persamaan. Kami menetapkan suhu terbalik, β = 1/ kT , menjadi 1,0 mol kkal −1 untuk menetapkan bobot pada setiap titik yang merupakan perantara antara penyesuaian terhadap energi (β = 0,0 mol kkal −1 , yaitu bobot seragam) dan penyesuaian terhadap populasi Boltzmann pada suhu kamar (β ∼1,7 mol kkal −1 ). Pilihan kami terhadap β, yang sesuai dengan suhu 500 K (β = 1/ kT ), memastikan tingkat akurasi yang tinggi pada minimum profil energi tanpa menimbulkan kesalahan besar di daerah penghalang. Energi medan gaya, E MM , diberikan oleh energi Amber ff99SB, E A99SB , ditambah istilah torsi baru, yang menggantikan torsi Amber ff99SB yang ada, V A99SB (θ):
Dalam persamaan ini, k 0 adalah konstanta, k m s adalah parameter dari kecocokan dan mewakili konstanta gaya untuk suku-suku M dalam perluasan kosinus, dan θ 0 ditetapkan ke 0,0°, konsisten dengan konvensi medan gaya Amber. Diformulasikan dengan cara ini, parameter yang dihasilkan mendefinisikan potensi torsi baru yang dimaksudkan untuk menggantikan suku torsi yang ada, V A99SB (θ), dalam ff99SB. Jumlah suku, M , yang digunakan dalam perluasan kosinus adalah dua untuk Ile, tiga untuk Leu, dan enam untuk χ 1 dan χ 2 dalam Asp dan Asn. Memperbolehkan lebih banyak parameter dalam torsi Ile dan Leu tidak menghasilkan peningkatan substansial dari kecocokan kuadrat terkecil.
HASIL
Perbandingan distribusi rotamer dari simulasi MD dengan statistik PDB
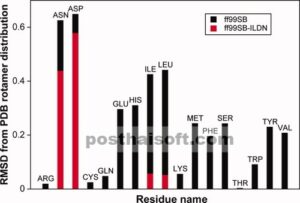
Pendekatan kami untuk penyempurnaan medan gaya Amber ff99SB difokuskan pada peningkatan deskripsi sudut torsi rantai samping χ 1 . Penelitian sebelumnya telah menunjukkan bahwa distribusi struktur dalam PDB mungkin merupakan perkiraan yang baik untuk distribusi yang diharapkan diamati dalam simulasi MD. 8 , 34 – 36 Namun, kesepakatan tersebut tidak diharapkan cukup kuantitatif untuk membuat parameter medan gaya, dan sebagai gantinya kami menggunakan perbandingan antara statistik MD dan PDB hanya untuk mengidentifikasi residu yang rantai sampingnya mungkin dijelaskan secara tidak akurat oleh medan gaya. Lebih khusus lagi, kami melakukan serangkaian simulasi MD dari peptida heliks pendek dengan urutan (Ala) 4 Xaa (Ala) 4 , di mana Xaa adalah 1 dari 20 asam amino selain Gly, Ala, dan Pro. Dari simulasi ini, kami menghitung populasi relatif dari rotamer χ 1 plus (p), minus (m) dan trans (t) untuk setiap residu dan membandingkannya dengan populasi relatif yang diamati untuk residu yang sama dalam heliks di PDB 13 (Gbr. 1 ; lihat juga Informasi Pendukung). Perbandingan ini menunjukkan dengan jelas bahwa distribusi χ 1 untuk empat residu (Ile, Leu, Asp, dan Asn) berbeda secara signifikan dari yang ditemukan dalam PDB. Kami menemukan bahwa hasil ini kuat sehubungan dengan metrik kesamaan yang digunakan untuk membandingkan distribusi dan panjang simulasi. Atas dasar pengamatan ini, kami memilih keempat residu ini untuk penyempurnaan parameter torsi χ 1 , seperti yang dijelaskan di bawah ini. Kami mencatat di sini bahwa perbandingan berikutnya antara simulasi protein dan pengukuran NMR, seperti yang dijelaskan lebih lanjut di bawah ini, menemukan keempat residu yang sama paling menyimpang.
Penyesuaian potensi torsi dengan energi yang dihitung QM
Perhitungan ab initio tingkat kuantum pada tingkat teori DF-LMP2 digunakan untuk menghitung profil energi torsi untuk empat asam amino (Gbr. 2 dan Informasi Pendukung). Karena Asp dan Asn menampilkan preferensi rotamerik yang lebih rumit untuk χ 2 daripada Ile dan Leu, dan karena torsi χ 1 dan χ 2 tampaknya lebih kuat digabungkan dalam Asp dan Asn daripada dalam Ile dan Leu, kami menghitung profil energi χ 1 / χ 2 dua dimensi penuh untuk Asp dan Asn dan menyesuaikan data QM yang dihasilkan ke profil torsi baru untuk χ 1 dan χ 2 . Untuk Ile dan Leu, kami menghitung pemindaian torsi QM untuk χ 1 pada tiga nilai χ 2 . Meskipun alasan fisik yang mendasari perbedaan antara QM dan energi medan gaya tidak jelas, kami memutuskan untuk mengikuti pendekatan yang digunakan untuk memodifikasi potensi tulang punggung ff99SB dengan memodifikasi istilah energi torsi dalam medan gaya. Modifikasi istilah terikat seperti untuk sudut torsi hanya akan secara langsung memengaruhi sejumlah kecil atom dan dengan demikian mengurangi kemungkinan menimbulkan efek samping yang tidak diinginkan (bila dibandingkan dengan, misalnya, modifikasi istilah tidak terikat).

Profil energi torsi untuk rotasi di sekitar sudut χ 1 dalam isoleusin. Profil energi dihitung untuk tiga nilai sudut χ 2 yang berbeda , yaitu ( A ) −60°, ( B ) 60°, dan ( C ) 178°. Sudut dihedral yang ditunjukkan di sini didefinisikan sebagai N–C α –C β –C γ2 . Energi ab initio yang dihitung pada level LMP2 dilaporkan dalam garis biru solid, sedangkan energi medan gaya Amber ff99SB dilaporkan dalam garis hitam solid. Istilah torsi yang dimodifikasi untuk sudut χ 1 (lihat Tabel I ) diperkenalkan untuk memaksimalkan kesesuaian antara energi ab initio dan energi medan gaya. Energi Amber ff99SB-ILDN yang dihasilkan dilaporkan dalam garis merah solid. Garis putus-putus menunjukkan perbedaan antara energi QM dan mekanika molekuler (dengan ff99SB dalam garis putus-putus hitam dan ff99SB-ILDN dalam garis putus-putus merah).
Dalam studi sebelumnya, parameter torsi medan gaya telah dioptimalkan terhadap kalkulasi kimia kuantum menggunakan serangkaian fungsi target. 7 , 8 , 37 Pilihan fungsi target dapat memengaruhi medan gaya yang dihasilkan karena secara implisit dapat menimbang berbagai wilayah permukaan energi secara berbeda. Dalam kalkulasi kami, kami menemukan bahwa tidak mungkin untuk memperoleh akurasi sub-kkal mol −1 pada kecocokan dengan seluruh profil energi potensial hanya dengan memperkenalkan istilah torsi tambahan. Karena alasan ini, kecocokan langsung dengan profil energi, sementara memberikan kecocokan yang baik dengan wilayah penghalang rotasi, menghasilkan kesalahan yang tidak dapat diterima dalam populasi rotamer relatif. Di sisi lain, kecocokan dengan populasi suhu kamar memperkenalkan kesalahan substansial hingga beberapa kkal mol −1 di wilayah penghalang, karena wilayah ini memiliki faktor Boltzmann yang dapat diabaikan pada suhu kamar. Seperti dijelaskan lebih rinci di bagian Bahan dan Metode , kami telah mengadopsi pendekatan antara dari pemasangan kuadrat terkecil untuk populasi Boltzmann pada 500 K. Kami menemukan bahwa pilihan ini memastikan, dalam praktiknya, bahwa bobot daerah berenergi tinggi, meskipun lebih kecil, tidak sepenuhnya dapat diabaikan. Ini juga menghasilkan keseimbangan yang baik antara kebutuhan untuk mendapatkan populasi kesetimbangan yang baik (kesalahan residual di daerah ini biasanya <0,5 kkal mol −1 ) dan hambatan torsi yang wajar (kesalahan pada hambatan biasanya antara 0,5 dan 2 kkal mol −1 ) (Gbr. 2 dan Informasi Pendukung). Koreksi torsi χ 1 dapat diperkenalkan pada beberapa set atom yang semuanya, jika tidak ada fluktuasi panjang dan sudut ikatan, akan terkait dengan simetri rotasi. Namun, karena kami memutuskan untuk mengikuti konvensi dalam medan gaya Amber untuk memperbaiki pergeseran fase, θ 0 , menjadi nol, simetri ini rusak dalam hal istilah torsi yang dihasilkan. Untuk setiap koreksi χ 1 yang kami pertimbangkan, kami kemudian menyesuaikannya, satu per satu, baik sudut dihedral N–C α –C β –C γ maupun C′–C α –C β –C γ dan memilih salah satu yang menghasilkan kecocokan terbaik dengan data ab initio . Hasilnya adalah N–C α –C β –C γ2 untuk Ile, N–C α –C β –C γ untuk Asp, dan C′–C α –C β –C γ untuk Leu dan Asn. Menambahkan koreksi ke lebih dari satu sudut torsi yang mendefinisikan χ 1, atau, ekuivalennya, membiarkan fase menjadi bukan nol, ternyata dalam praktiknya hanya menghasilkan peningkatan sederhana dalam kualitas kecocokan, dan dengan demikian istilah torsi hanya untuk satu sudut dimodifikasi. Koreksi yang diperkenalkan dengan prosedur ini dilaporkan dalam Tabel I dan berkisar dari ∼1 kkal mol −1 untuk Leu hingga ∼5 kkal mol −1 untuk Asp. Kami menyebut medan gaya yang dihasilkan dari penggantian istilah dihedral asli dalam ff99SB dengan parameter yang dioptimalkan ini “ff99SB-ILDN” (ILDN adalah kode satu huruf untuk rantai samping yang potensinya kami modifikasi).
Tabel I. Daftar Parameter Modifikasi untuk Potensi Torsi χ 1 dan χ 2 pada Asam Amino Tertentu dari Medan Gaya Amber ff99SB
Res. Sudut θ 0 k 1 k 2 k 3 k 4 k 5 bahasa inggris 6
Pulau N–C α – C β –C γ2 0.0 0,195 -0,846
Singa C– Cα – Cβ – Cγ 0.0 0,571 tahun -0,358 0,135
Asp N–C α – C β –C γ 0.0 -2,635 -1.190 -0,007 0.423 0.232 -0,213
C α –C β –C γ –O δ1,2 0.0 0.0 -0,443 0.0 -0,138 0.0 -0,013
Asn C– Cα – Cβ – Cγ 0.0 0,571 tahun -0,596 0.118 -0,417 0,104 tahun -0,101
Cα – Cβ – Cγ – Nδ 0.0 -1,046 -0,181 -0,035 0.100 0.130 -0,106
Parameternya dalam kkal mol −1 dan sesuai dengan potensi torsi yang didefinisikan dalam teks utama. Perhatikan bahwa untuk torsi χ 2 dalam Asp, koreksi diterapkan pada kedua atom oksigen rantai samping.
Distribusi rotamer dalam medan gaya ff99SB-ILDN
Sebagai uji pertama, kami mengulangi simulasi peptida heliks pendek menggunakan potensi torsi rantai samping yang dimodifikasi. Untuk keempat residu yang kami modifikasi, kami menemukan bahwa medan gaya ff99SB-ILDN meningkatkan kesesuaian dengan distribusi PDB (Gbr. 1 ; lihat juga Informasi Pendukung). Hasil ini menggembirakan karena informasi tentang distribusi PDB tidak digunakan dengan cara apa pun untuk memodifikasi parameter torsi. Kami mengamati peningkatan substansial untuk Leu dan Ile dan peningkatan marjinal untuk Asp dan Asn. Alasan yang mendasari perbedaan ini tidak jelas. Meskipun medan gaya yang “sempurna” tidak akan selalu mereproduksi distribusi PDB secara tepat, penyimpangan yang diamati untuk Asp dan Asn dapat disebabkan oleh kesalahan dalam parameterisasi interaksi yang tidak terikat. Kesalahan tersebut tidak dapat sepenuhnya dikompensasi atau dikoreksi dengan memperkenalkan potensi torsi yang dimodifikasi. Namun, seperti yang dijelaskan dalam bagian berikut, kami mengamati peningkatan substansial untuk keempat jenis residu ketika ff99SB dan ff99SB-ILDN dievaluasi menggunakan pengukuran NMR keadaan larutan.
Validasi melalui perbandingan dengan data NMR
Pencocokan distribusi rotamer PDB bukanlah kontrol langsung yang dapat digunakan untuk mengevaluasi kualitas medan gaya. Uji yang lebih penting dan ketat adalah kemampuan medan gaya untuk mereproduksi kuantitas eksperimental yang secara langsung menyelidiki konformasi rantai samping protein dalam larutan. Sejumlah besar data eksperimental tersebut tersedia dalam bentuk kopling skalar rantai samping NMR 3 J dan RDC. Kami telah melakukan simulasi MD pada skala waktu mikrodetik untuk empat protein globular (BPTI, ubiquitin, GB3, dan lisozim) di mana sejumlah besar data NMR berkualitas tinggi yang menyelidiki konformasi rantai samping tersedia. Simulasi yang relatif lama diperlukan untuk mencapai konvergensi yang kuat dari kuantitas NMR yang dihitung, karena perubahan rotamerik terjadi pada rentang skala waktu yang luas. Simulasi dilakukan dengan menggunakan medan gaya ff99SB standar dan medan gaya ff99SB-ILDN yang dimodifikasi. Untuk masing-masing dari keempat protein, kami menemukan keadaan asli sangat stabil dalam simulasi dengan ff99SB dan ff99SB-ILDN sebagaimana dibuktikan, misalnya, oleh deviasi akar-rata-kuadrat (RMSD) tulang punggung rata-rata yang rendah dari struktur yang ditentukan secara eksperimen (≤1 Å dalam simulasi BPTI, Ubq, dan GB3 dan <2 Å dalam simulasi HEWL).
Kami menghitung sejumlah besar kopling skalar dari simulasi ini dan membandingkannya dengan nilai eksperimen; hasil untuk Ile, Leu, Asp, dan Asn ditunjukkan pada Gambar 3. Jelas bahwa banyak outlier yang ada dalam simulasi dengan ff99SB tidak ada dalam simulasi dengan ff99SB-ILDN. Untuk mengukur kesesuaian antara eksperimen dan simulasi, kami menghitung RMSD antara kopling skalar yang diperoleh secara eksperimen dan simulasi berdasarkan per jenis residu. Hasilnya ditunjukkan pada Gambar 4 , di mana nilai RMSD ini diplot terhadap hasil yang diperoleh dari analisis peptida heliks yang dijelaskan di atas. Hasil untuk ff99SB menunjukkan dengan jelas bahwa keempat residu yang dipilih untuk penyempurnaan medan gaya berdasarkan perbandingan antara distribusi rotamer dalam PDB dan dalam simulasi MD peptida heliks juga merupakan residu yang menampilkan deviasi terbesar antara data NMR yang dihitung dan eksperimen. Pengamatan ini memvalidasi pendekatan kami dalam menggunakan deviasi dari distribusi rotamer PDB dalam heliks sebagai metrik untuk mengidentifikasi residu yang memerlukan penyempurnaan parameter torsi rantai sampingnya. Sesuai dengan pemeriksaan visual Gambar 3 , hasil pada Gambar 4 menunjukkan bahwa deskripsi keempat residu yang dimodifikasi dalam ff99SB-ILDN meningkat secara signifikan setelah penyempurnaan. Khususnya, untuk Asp dan Asn, kesesuaian dengan data NMR meningkat jauh lebih banyak daripada kesesuaian dengan distribusi rotamer PDB.
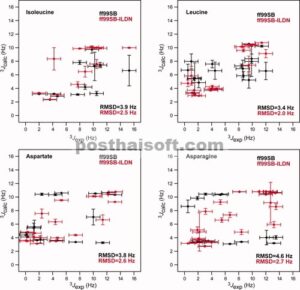
Perbandingan kopling skalar NMR 3 J eksperimental dan nilai-nilai terkait yang dihitung dari simulasi MD HEWL, BPTI, Ubq, dan GB3. Plot menunjukkan kopling H α –C α –C β –H β1,2 yang secara langsung menguji sudut χ 1 rantai samping . Nilai sebelum (hitam) dan setelah (merah) penyempurnaan potensi torsi rantai samping dilaporkan untuk empat residu (Ile, Leu, Asp, dan Asn) yang potensi rantai sampingnya dimodifikasi. Setiap panel diberi label dengan RMSD antara kopling skalar eksperimental, dan nilai-nilai yang dihitung menggunakan dua medan gaya.
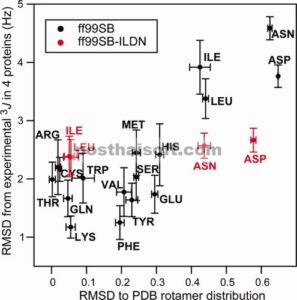
Perbandingan berbagai metrik yang digunakan untuk mengevaluasi kualitas deskripsi rantai samping dalam medan gaya. RMSD antara distribusi rotamer yang dihitung dan distribusi yang diamati dalam PDB diplot versus RMSD antara kopling NMR rantai samping yang dihitung dan eksperimental 3 J (H α –C α –C β –H β1,2 ). Nilai yang dihitung setelah penyempurnaan potensi torsi rantai samping dilaporkan dalam warna merah.
Kami juga membandingkan simulasi dengan RDC yang diukur secara eksperimental yang bertindak sebagai probe alternatif dari orientasi rantai samping asam amino. Kami menghitung RDC untuk ikatan C β –H β1,2 dalam GB3 dan membandingkannya dengan nilai eksperimental [Gbr. 5 (a)]. Kami mengamati peningkatan untuk residu Asp dan Asn, meskipun perbandingan ini rumit karena penugasan stereospesifik tidak tersedia. Untuk HEWL, kami membandingkan simulasi kami dengan RDC yang diukur secara eksperimental untuk gugus amida rantai samping dalam residu Asn [Gbr. 5 (b)]. Nilai-nilai ini bergantung pada torsi χ 1 dan χ 2 dan juga menunjukkan peningkatan signifikan dalam medan gaya ff99SB-ILDN.
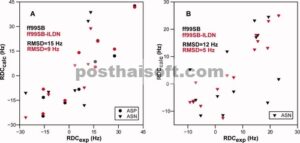
Perbandingan kopling dipolar residual eksperimental dan nilai yang dihitung dari simulasi MD ( A ) GB3 dan ( B ) HEWL. Dalam A, kami menunjukkan perbandingan antara eksperimen dan simulasi untuk C β –H β1,2 RDCs di GB3 untuk residu Asp (lingkaran) dan Asn (segitiga). Dalam B, kami menunjukkan perbandingan antara eksperimen dan simulasi untuk N δ –H δ1,2 RDCs di HEWL untuk residu Asn (segitiga). Nilai eksperimental dilaporkan sebagai jumlah kopling ke dua proton amida rantai samping, sehingga kami menghitung jumlah yang sama dari simulasi MD. Dalam A dan B, hasil ditunjukkan untuk kedua simulasi dengan ff99SB (simbol hitam) dan dengan ff99SB-ILDN (simbol merah). Setiap panel diberi label dengan RMSD antara kopling eksperimental, dan nilai dihitung menggunakan dua medan gaya.
DISKUSI
Kami mengusulkan serangkaian parameter torsi rantai samping yang ditingkatkan untuk medan gaya Amber ff99SB. Penyempurnaan dibatasi pada empat residu (isoleusin, leusin, aspartat, dan asparagin) yang tampaknya paling bermasalah dalam ff99SB saat membandingkan distribusi rotamer yang diamati dalam simulasi MD heliks pendek dengan distribusi rotamer yang diambil dari heliks dalam PDB. Parameter baru diperoleh dengan menyesuaikan data QM baru dan divalidasi terhadap serangkaian besar data NMR. Peningkatan konsisten yang diamati untuk keempat residu yang kami modifikasi menunjukkan bahwa pendekatan kami kuat dan umum. Meskipun demikian, kami memutuskan untuk tidak memodifikasi residu tambahan, karena akan semakin sulit untuk menunjukkan peningkatan yang signifikan untuk residu tersebut. Koreksi yang diperkenalkan di sini untuk Ile, Leu, Asp, dan Asn berkisar antara 1 dan 5 kkal mol −1 dan dengan demikian dapat memiliki dampak yang nyata pada stabilitas lipatan protein dan loop fleksibel, khususnya dalam simulasi MD panjang yang melampaui rentang waktu rotasi rantai samping yang terkubur atau terkubur sebagian. Karena potensi torsi baru yang dijelaskan di sini merupakan peningkatan yang jelas dari potensi torsi di medan gaya yang ada dan tampaknya tidak menyebabkan efek samping yang tidak diinginkan, kami merekomendasikan penggunaan ff99SB-ILDN daripada ff99SB dalam simulasi MD protein.
Ucapan Terima Kasih
Penulis mengucapkan terima kasih kepada semua peneliti eksperimental yang datanya telah mereka gunakan dan tanpa mereka penelitian ini tidak akan mungkin terlaksana. Secara khusus, mereka mengucapkan terima kasih kepada Ad Bax dan John L. Marquardt karena telah membagikan data yang belum dipublikasikan tentang ubiquitin dan Christina Redfield dan Lorna Smith karena telah membagikan data tentang lisozim. Mereka juga mengucapkan terima kasih kepada Michael Eastwood karena telah membaca naskah dengan saksama dan Rebecca Kastleman atas bantuan penyuntingannya.